Learning outcomes
- Relate disorders of globin change synthesis to the pathogenesis of thalassaemia and haemoglobin variants
- Describe the pathogenesis and clinical presentation of alpha-thalassaemia
- Predict the risk of thalassaemia based on the genetic inheritance pattern of alpha-thalassaemia
- List common molecular mutations associated with alpha-thalassaemia in Malaysia
- Appraise the laboratory tests that are commonly used for screening and diagnosis of thalassaemia and haemoglobin variants
Prevalence
Alpha thalassaemia is among the most common monogenic inherited disorder worldwide. It is estimated that about 5% of the population worldwide carry an 𝜶-thalassaemia mutation. In Malaysia, 4.1% of secondary school students carry an 𝜶-globin defect.
Molecular basis of 𝜶-thalassaemia
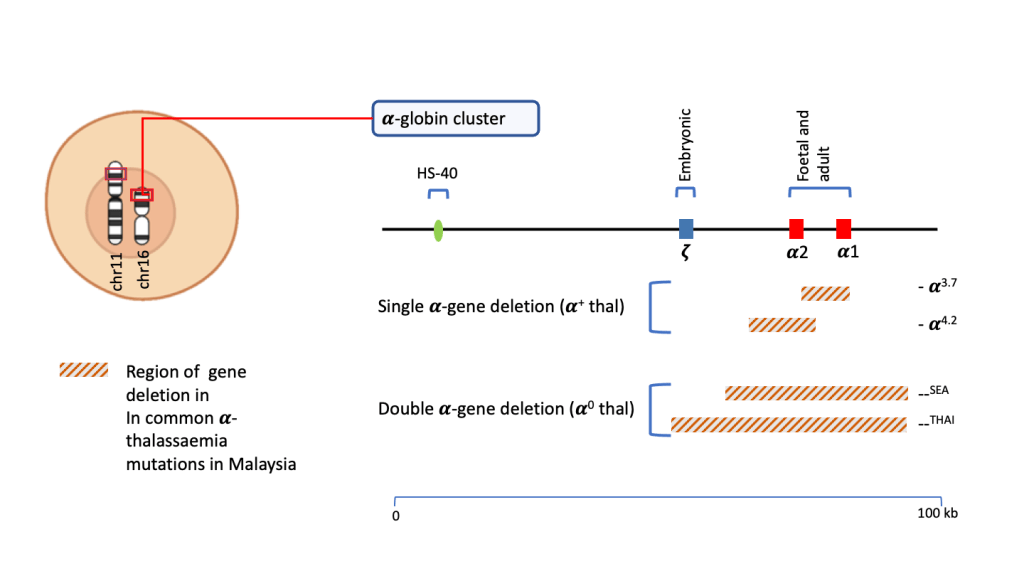
𝜶-thalassaemia results from mutations in any of the four 𝜶-globin genes (HBA1, HBA2) that are present in an individual. Individuals inherit one pair of genes from the male partner and the other pair from the female partner, giving a total of four 𝜶-alleles in the individual.
The majority of mutations that cause 𝜶-thalassaemia are considered as deletion mutations where large portion of the HBA1 and/or HBA2genes are deleted. In Malaysia, the common mutations causing 𝜶-thalassaemia are;
- —SEA
- —THAI
- -𝜶3.7
- -𝜶4.2
Both —SEA and —THAI cause deletions of both the HBA1 and HBA2 genes while -𝜶3.7 and -𝜶4.2 result in deletion of only one of either genes. Carriers of -𝜶3.7 is most prevalent among Malays, native Sabahans and Indians while —SEA is more prevalent in Chinese.
Clinical and laboratory features
The clinical presentation of 𝜶-thalassaemia is reflected by the number of 𝜶-globin alleles that is deleted as a result of a mutation. The mildest form is 𝜶-thalassaemia when only a single allele is deleted (-𝜶/𝜶𝜶) while the severest form is Hb Barts disease where all four 𝜶-alleles are deleted (–/–).

𝜶-thalassaemia silent carrier (-𝜶/𝜶𝜶)
This condition occurs when the individual is heterozygous for 𝜶+-thal mutations that effects only a single gene such as -𝜶3.7 or -𝜶4.2. Such individuals are completely asymptomatic and have normal haemoglobin and red cell parameters including MCV and MCH. They are usually identified on DNA molecular studies especially when they are investigated as part of a family study.
𝜶-thalassaemia minor or trait (–/𝜶𝜶 or -𝜶/-𝜶)
Individuals with 𝜶-thalassaemia trait are usually identified incidentally during a full blood count. They are generally asymptomatic with mild hypochromic microcytic anaemia. MCV and MCH is low, with MCH usually less than 28pg. The red blood count (RBC) is usually normal or high which is in contrast with iron deficiency where the RBC is low. Red cell anisocytosis is also a not a prominent feature in 𝜶-thalassaemia trait as evidenced by normal RDW. Confirmation of the diagnosis will require DNA analysis of the 𝜶-globin gene.
The clinical and laboratory features are indistinguishable between heterozygous 𝜶0-thal (–/𝜶𝜶) and homozygous 𝜶+ (-𝜶/-𝜶). Distinction between the two is only possible by DNA analysis, which may be necessary for genetic counselling when the partner is also identified as 𝜶-thalassaemia trait.
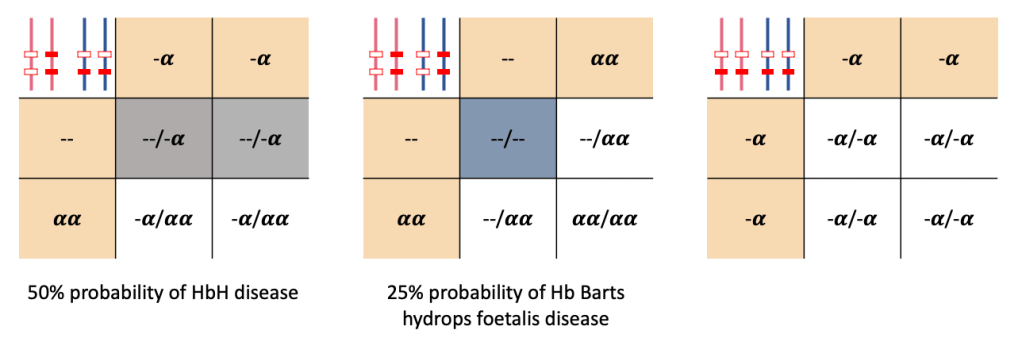
Some of the potential scenarios of outcome of pregnancy from two partners that are 𝜶-thalassaemia trait are illustrated below using Punnett squares.
HbH disease (–/-𝜶)
As illustrated above, HbH disease occurs with loss of three 𝜶-genes. Family studies would indicate that one parent is heterozygous for 𝜶0-thal with the other parent heterozygous for 𝜶+-thal.
The presence of only a single remaining 𝜶-globin gene, results in markedly reduced production of 𝜶-globin. This leads to an imbalance of 𝜶/𝜷-globin chain ratio. In the absence of 𝜶-globin chains, the 𝜷-globin chains in adults combine among themselves to for 𝜷-tetramers which is termed HbH (𝜷4). HbH is unstable and is readily denatured in the red cells, resulting in cell damage and premature destruction in the spleen.
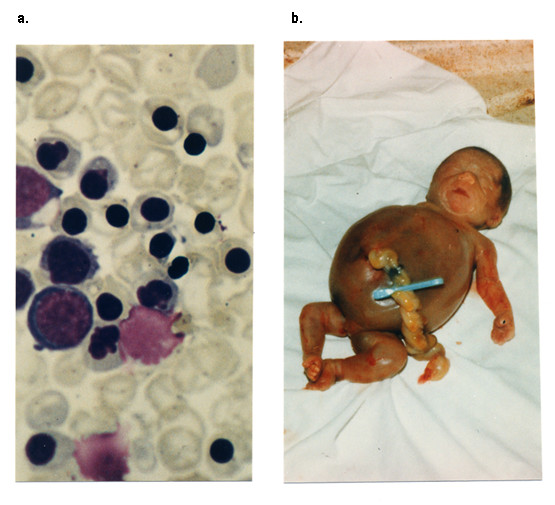
HbH containing red cells can be demonstrated by treating the cells with a supravital dye that causes HbH to precipitate, giving a characteristic ‘golf-ball’ pattern when viewed microscopically.
The clinical presentation of HbH disease is variable. Some patients present with mild to moderate anaemia and splenomegaly without requiring blood transfusion, while others may have a more severe course with presentation early in life requiring blood transfusion support. The more severe forms may be associated with increasing splenomegaly, iron overload, recurrent infections, gallstones and megaloblastic anaemia secondary to folate deficiency.
Hb Barts hydrops foetalis
Hb Barts hydros foetal is is the most severe form of 𝜶-thalassaemia in which all four of the 𝜶-globin alleles are lost. Both parents would be expected to be heterozygous for an 𝜶0-thal mutation.
This condition is incompatible with life as 𝜶-globin chains are necessary for formation of both foetal HbF (𝜶2𝜸2) and adult HbA (𝜶2𝜷2). In the absence of any 𝜶-globin, the 𝜸-globin chains in the foetus combines to form non-functioning 𝜸-tetramers (𝜸4) which is called Hb Barts.
The early part of embryonal development may be sustained by presence of embryonic haemoglobin such as 𝜻2𝜸2 and 𝜻2𝜷2. However, in later developmental stages, the foetus develops severe anaemia in the absence of HbF. Compensatory increase in cardiac output leads to congestive cardiac failure with generalised foetal oedema and enlarged placenta, giving the characteristic picture of hydrops foetalis on ultrasound and gross examination. The foetus either dies in-utero or is delivered as a stillbirth.