Learning outcome
- Describe the pathophysiology, inheritance, clinical and laboratory features of von Willebrand disease
Introduction
The coagulation cascade consists of many interacting proteins that serve as activating proteases and co-factors that amplify the reaction. Functional mutations have been identified in nearly all of the proteins within the coagulation cascade, that results in an heritable bleeding disorder.
The more common congenital bleeding disorders that is observed include;
- von Willebrand Factor deficiency or dysfunction causing von Willebrand disease (vWD)
- FVIII deficiency causing Haemophilia A
- FIX deficiency causing Haemophilia B
Other bleeding disorders such as deficiencies of FXI, FVII and FXIII and fibrinogen have been reported in Malaysia, but they are extremely rare.
Von Willebrand disease (vWD)
Pathophysiology
Von Willebrand disease is a clinically heterogenous group of bleeding disorders that occurs secondary to deficiency of production or production of abnormal vWF. It is the commonest of the inherited coagulation disorders with up to 1% of the population effected. No significant geographical variation in its prevalence is noted.
Many subtypes of vWD have been recognised. The majority of cases are inherited in an autosomal dominant fashion, with equal representation of male and female.
As described in the earlier section, vWF is especially important in the initiation of clotting. It is produced by endothelial cells and megakaryocytes /platelets as ultra-large multimers, which are then processed into shorter multimers by ADAMTS13. The vWF protein contains multiple domains that interact with platelets, collagen and coagulation factors.
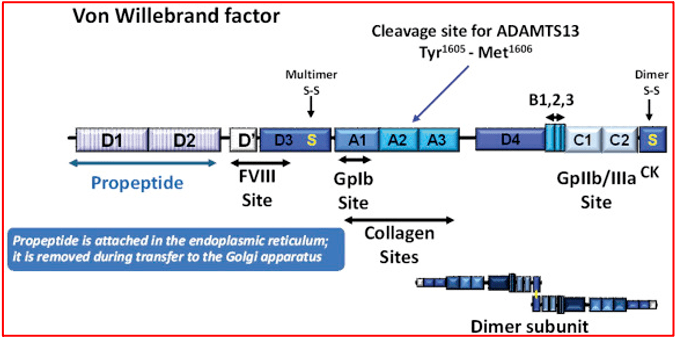
In addition, it serves as a carrier protein for FVIII in circulation and prevents its degradation. At the site of vascular injury, vWF releases the FVIII on action of thrombin, to partake in the formation of the ‘thrombin burst’. Deficiency of vWF therefore is usually associated with concomitant FVIII deficiency.
Clinical features
In most cases, bleeding associated with vWD is mild and limited to skin (easy bruising) or mucosal bleeds (epistaxis, bleeding gums). Women may present with troublesome menorrhagia. Some patients may present with prolonged bleeding following a dental procedure. Deep bleeds e.g. intra-articular bleeds are uncommon which is in contrast to Haemophilia A.
Laboratory features
Patients with vWD usually have a normal platelet count. An exception may be seen in the rare Type IIB vWD, where thrombocytopenia may be observed as a consequence of vWF overactivity and adhesion of platelets to the activated vWF.
An isolated prolongation of APTT may be observed with normal PT and TT. The APTT prolongation may be mild and multiple repeated testing may be required before a definitive diagnosis is made. Studies of vWF levels and activity (vWF.Ag, vWF.Activity, vWF.RiCoF, multiuser analysis) would be required to confirm the diagnosis and determine the vWD subtype. FVIII levels are also reduced.
Management
Mild vWD can be treated with local measures such as pressure application and nasal packing for epistaxis. Tranexamic acid, a drug used to prevent breakdown of formed clots is useful adjunctive treatment.
Mild subtypes of vWD (Type I, Type IIA) may be managed with DDAVP. DDAVP stimulates release of vWF from endothelial cell stores and effects a transient increase of vWF in circulation. Factor concentrates containing vWF or cryoprecipitate would be needed If sustained high levels of vWF are required as in severe bleeds or in post-operative management.
